Regulatory Writing: An Overview
Third Edition
7 Regulatory Affairs Professionals Society
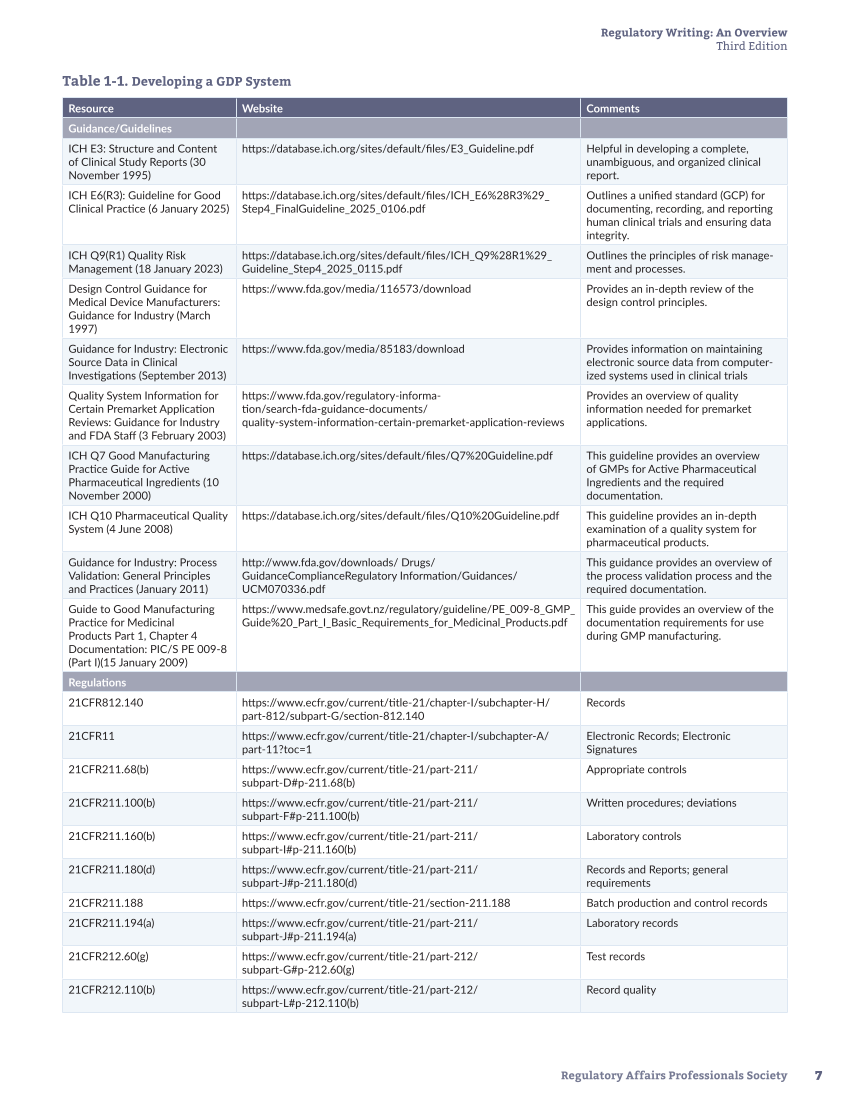
Table 1-1. Developing a GDP System
Resource Website Comments
Guidance/Guidelines
ICH E3: Structure and Content
of Clinical Study Reports (30
November 1995)
https://database.ich.org/sites/default/files/E3_Guideline.pdf Helpful in developing a complete,
unambiguous, and organized clinical
report.
ICH E6(R3): Guideline for Good
Clinical Practice (6 January 2025)
https://database.ich.org/sites/default/files/ICH_E6%28R3%29_
Step4_FinalGuideline_2025_0106.pdf
Outlines a unified standard (GCP) for
documenting, recording, and reporting
human clinical trials and ensuring data
integrity.
ICH Q9(R1) Quality Risk
Management (18 January 2023)
https://database.ich.org/sites/default/files/ICH_Q9%28R1%29_
Guideline_Step4_2025_0115.pdf
Outlines the principles of risk manage-
ment and processes.
Design Control Guidance for
Medical Device Manufacturers:
Guidance for Industry (March
1997)
https://www.fda.gov/media/116573/download Provides an in-depth review of the
design control principles.
Guidance for Industry: Electronic
Source Data in Clinical
Investigations (September 2013)
https://www.fda.gov/media/85183/download Provides information on maintaining
electronic source data from computer-
ized systems used in clinical trials
Quality System Information for
Certain Premarket Application
Reviews: Guidance for Industry
and FDA Staff (3 February 2003)
https://www.fda.gov/regulatory-informa-
tion/search-fda-guidance-documents/
quality-system-information-certain-premarket-application-reviews
Provides an overview of quality
information needed for premarket
applications.
ICH Q7 Good Manufacturing
Practice Guide for Active
Pharmaceutical Ingredients (10
November 2000)
https://database.ich.org/sites/default/files/Q7%20Guideline.pdf This guideline provides an overview
of GMPs for Active Pharmaceutical
Ingredients and the required
documentation.
ICH Q10 Pharmaceutical Quality
System (4 June 2008)
https://database.ich.org/sites/default/files/Q10%20Guideline.pdf This guideline provides an in-depth
examination of a quality system for
pharmaceutical products.
Guidance for Industry: Process
Validation: General Principles
and Practices (January 2011)
http://www.fda.gov/downloads/ Drugs/
GuidanceComplianceRegulatory Information/Guidances/
UCM070336.pdf
This guidance provides an overview of
the process validation process and the
required documentation.
Guide to Good Manufacturing
Practice for Medicinal
Products Part 1, Chapter 4
Documentation: PIC/S PE 009-8
(Part I)(15 January 2009)
https://www.medsafe.govt.nz/regulatory/guideline/PE_009-8_GMP_
Guide%20_Part_I_Basic_Requirements_for_Medicinal_Products.pdf
This guide provides an overview of the
documentation requirements for use
during GMP manufacturing.
Regulations
21CFR812.140 https://www.ecfr.gov/current/title-21/chapter-I/subchapter-H/
part-812/subpart-G/section-812.140
Records
21CFR11 https://www.ecfr.gov/current/title-21/chapter-I/subchapter-A/
part-11?toc=1
Electronic Records Electronic
Signatures
21CFR211.68(b) https:/ www.ecfr.gov/ current/ title-21/ part-211/
subpart-D#p-211.68(b)
Appropriate controls
21CFR211.100(b) https:/ www.ecfr.gov/ current/ title-21/ part-211/
subpart-F#p-211.100(b)
Written procedures deviations
21CFR211.160(b) https:/ www.ecfr.gov/ current/ title-21/ part-211/
subpart-I#p-211.160(b)
Laboratory controls
21CFR211.180(d) https:/ www.ecfr.gov/ current/ title-21/ part-211/
subpart-J#p-211.180(d)
Records and Reports general
requirements
21CFR211.188 https://www.ecfr.gov/current/title-21/section-211.188 Batch production and control records
21CFR211.194(a) https:/ www.ecfr.gov/ current/ title-21/ part-211/
subpart-J#p-211.194(a)
Laboratory records
21CFR212.60(g) https:/ www.ecfr.gov/ current/ title-21/ part-212/
subpart-G#p-212.60(g)
Test records
21CFR212.110(b) https:/ www.ecfr.gov/ current/ title-21/ part-212/
subpart-L#p-212.110(b)
Record quality
Third Edition
7 Regulatory Affairs Professionals Society
Table 1-1. Developing a GDP System
Resource Website Comments
Guidance/Guidelines
ICH E3: Structure and Content
of Clinical Study Reports (30
November 1995)
https://database.ich.org/sites/default/files/E3_Guideline.pdf Helpful in developing a complete,
unambiguous, and organized clinical
report.
ICH E6(R3): Guideline for Good
Clinical Practice (6 January 2025)
https://database.ich.org/sites/default/files/ICH_E6%28R3%29_
Step4_FinalGuideline_2025_0106.pdf
Outlines a unified standard (GCP) for
documenting, recording, and reporting
human clinical trials and ensuring data
integrity.
ICH Q9(R1) Quality Risk
Management (18 January 2023)
https://database.ich.org/sites/default/files/ICH_Q9%28R1%29_
Guideline_Step4_2025_0115.pdf
Outlines the principles of risk manage-
ment and processes.
Design Control Guidance for
Medical Device Manufacturers:
Guidance for Industry (March
1997)
https://www.fda.gov/media/116573/download Provides an in-depth review of the
design control principles.
Guidance for Industry: Electronic
Source Data in Clinical
Investigations (September 2013)
https://www.fda.gov/media/85183/download Provides information on maintaining
electronic source data from computer-
ized systems used in clinical trials
Quality System Information for
Certain Premarket Application
Reviews: Guidance for Industry
and FDA Staff (3 February 2003)
https://www.fda.gov/regulatory-informa-
tion/search-fda-guidance-documents/
quality-system-information-certain-premarket-application-reviews
Provides an overview of quality
information needed for premarket
applications.
ICH Q7 Good Manufacturing
Practice Guide for Active
Pharmaceutical Ingredients (10
November 2000)
https://database.ich.org/sites/default/files/Q7%20Guideline.pdf This guideline provides an overview
of GMPs for Active Pharmaceutical
Ingredients and the required
documentation.
ICH Q10 Pharmaceutical Quality
System (4 June 2008)
https://database.ich.org/sites/default/files/Q10%20Guideline.pdf This guideline provides an in-depth
examination of a quality system for
pharmaceutical products.
Guidance for Industry: Process
Validation: General Principles
and Practices (January 2011)
http://www.fda.gov/downloads/ Drugs/
GuidanceComplianceRegulatory Information/Guidances/
UCM070336.pdf
This guidance provides an overview of
the process validation process and the
required documentation.
Guide to Good Manufacturing
Practice for Medicinal
Products Part 1, Chapter 4
Documentation: PIC/S PE 009-8
(Part I)(15 January 2009)
https://www.medsafe.govt.nz/regulatory/guideline/PE_009-8_GMP_
Guide%20_Part_I_Basic_Requirements_for_Medicinal_Products.pdf
This guide provides an overview of the
documentation requirements for use
during GMP manufacturing.
Regulations
21CFR812.140 https://www.ecfr.gov/current/title-21/chapter-I/subchapter-H/
part-812/subpart-G/section-812.140
Records
21CFR11 https://www.ecfr.gov/current/title-21/chapter-I/subchapter-A/
part-11?toc=1
Electronic Records Electronic
Signatures
21CFR211.68(b) https:/ www.ecfr.gov/ current/ title-21/ part-211/
subpart-D#p-211.68(b)
Appropriate controls
21CFR211.100(b) https:/ www.ecfr.gov/ current/ title-21/ part-211/
subpart-F#p-211.100(b)
Written procedures deviations
21CFR211.160(b) https:/ www.ecfr.gov/ current/ title-21/ part-211/
subpart-I#p-211.160(b)
Laboratory controls
21CFR211.180(d) https:/ www.ecfr.gov/ current/ title-21/ part-211/
subpart-J#p-211.180(d)
Records and Reports general
requirements
21CFR211.188 https://www.ecfr.gov/current/title-21/section-211.188 Batch production and control records
21CFR211.194(a) https:/ www.ecfr.gov/ current/ title-21/ part-211/
subpart-J#p-211.194(a)
Laboratory records
21CFR212.60(g) https:/ www.ecfr.gov/ current/ title-21/ part-212/
subpart-G#p-212.60(g)
Test records
21CFR212.110(b) https:/ www.ecfr.gov/ current/ title-21/ part-212/
subpart-L#p-212.110(b)
Record quality